

Phoenix Defective Medical Device Lawyer
Written by: Hastings Law Firm | Reviewed by: Tommy Hastings | Updated: May 6, 2026
Defective medical devices can fail inside the body and leave patients facing painful complications, additional procedures, and lasting harm. Product liability focuses on whether a device was unreasonably dangerous due to a design defect, a manufacturing defect, or a failure to warn, and responsibility can extend beyond the manufacturer through the chain of distribution. FDA clearance and recalls can shape how these claims are evaluated, and preserving an explanted device can be critical to understanding what went wrong. If you or a loved one were harmed or worse due to a defective medical device in Phoenix, Arizona, contact Hastings Law Firm for a free, confidential case review.

Trusted Medical Product Liability Attorneys in Phoenix
What You Should Know About Medical Equipment Failure Claims in Phoenix:
- Recovery can depend on whether the harm is tied to a design defect, a manufacturing defect, or a failure to warn.
- Liability can extend beyond the manufacturer when other entities in the chain of distribution contributed to the harm.
- Options can be limited when manufacturers raise federal preemption defenses tied to FDA clearance.
- Compensation can include medical costs, lost wages, and pain and suffering when a defective device causes life altering complications.
- Wrongful death damages may be available when a defective device causes a patient death.
- Case outcomes can vary widely because many device claims are handled as individual lawsuits in multi district litigation rather than class actions.
- The ability to pursue a claim can be lost if filing deadlines are missed under Arizona law.
- Proving a defect can become significantly harder if the explanted device is returned to the manufacturer or destroyed.
- Causation disputes can drive outcomes when the injury could also be attributed to an underlying condition.
- Recall history and adverse event reporting data can be central when evaluating whether risks were known before injuries occurred.

A Healthcare Focused Law Firm
When a medical device fails inside your body, the consequences can be devastating. You trusted the device, the surgeon, and the system behind them. If that trust was broken, you deserve answers and a clear path forward. As a Phoenix defective medical device lawyer team, Hastings Law Firm focuses exclusively on medical negligence and product liability. Our legal team, which includes former defense attorneys and in-house medical professionals, investigates every link in the chain to determine who is responsible for your injury. If you or a loved one has been harmed by a defective implant or medical product, contact us for a free, confidential case evaluation. We work on a contingency fee basis, meaning you pay no attorney fees or costs unless we recover compensation for you.
Understanding Product Liability: How Medical Devices Fail Patients
A defective medical device claim is typically built on one of three theories of liability: a design defect, a manufacturing defect, or a marketing defect, commonly known as failure to warn. Each theory targets a different point in the product’s life cycle, and identifying the right one is central to building a strong case.
Product liability law holds manufacturers and other parties responsible when a medical product causes harm. Unlike a standard medical malpractice claim, which focuses on a doctor’s decisions, product liability focuses on the product itself. Under strict liability, a manufacturer can be held accountable for injuries caused by a defective device even without proof of carelessness or intent. The product simply had to be unreasonably dangerous. This legal standard ensures that the burden of safety rests on the creator of the product rather than the patient, removing the need to prove the manufacturer acted maliciously.
Here are the three core defect types and how they apply:
- Design defect: A design defect is a flaw existing in the product’s blueprint that makes the device inherently unsafe before it is ever manufactured. This flaw affects every unit produced. Metal-on-metal hip implants are a well-known example; the design itself caused metal particles to shed into surrounding tissue, leading to serious complications in thousands of patients.
- Manufacturing defect: A manufacturing defect is an error introduced during the production or assembly process. This means the design may have been sound, but something went wrong with a specific unit or batch. Contamination during production, improper sterilization, or flawed assembly can all introduce defects that make one device dangerous while others from the same line function as intended.
- Marketing defect (failure to warn): This theory applies when a manufacturer knew about risks associated with the device but did not adequately disclose those risks to doctors or patients. If warnings were incomplete, misleading, or absent, and a patient was injured as a result, the manufacturer may be liable.
A Phoenix defective medical device lawyer must determine which defect theory applies based on the evidence. In many cases, more than one theory is relevant. Our defective medical device attorneys work alongside medical experts to analyze the device, review clinical data, and identify exactly where the failure occurred.

High-Risk and Commonly Recalled Medical Devices
Numerous devices have faced FDA recalls due to high failure rates, including metal-on-metal hip replacements, transvaginal mesh, CPAP machines, and certain cardiac implants. These products were used in thousands of patients before safety concerns came to light.
A manufacturer or the FDA issues a recall to correct a defective device or to address a risk of injury. These recalls can range from minor labeling corrections to full market withdrawals. You can review current and past actions through the U.S. Food and Drug Administration’s Medical Device Recalls and Early Alerts database.
Implant failure, which refers to a device malfunctioning or breaking down inside the body, can cause pain, infection, tissue damage, or the need for revision surgery. The table below outlines some of the most commonly affected device categories.
| Device Type | Common Complication | Typical Injury |
|---|---|---|
| Metal-on-metal hip implants | Metallosis, component loosening | Tissue necrosis, chronic pain, revision surgery |
| Transvaginal mesh | Erosion through tissue | Infection, organ perforation, chronic pain |
| Hernia mesh | Adhesion, mesh migration | Bowel obstruction, reoperation |
| CPAP machines (e.g., Philips Respironics) | Toxic foam degradation | Respiratory damage, potential carcinogen exposure |
| Cardiac devices (pacemakers, defibrillators) | Lead fracture, battery failure | Cardiac arrest, electrical shock, emergency surgery |
| Knee and shoulder implants | Loosening, component wear | Bone loss, instability, revision surgery |
If you have experienced complications from any of these device categories, a medical device lawyer in Phoenix can evaluate whether your injury is connected to a known defect or recall. Some of these products were used off-label, meaning they were implanted for a purpose not specifically approved by the FDA, which can further strengthen a claim.
The Hastings Law Firm Difference
Results matter, but what truly sets us apart is how we achieve them. Every verdict, every settlement, and every Phoenix courtroom victory comes from one guiding promise: To treat each client’s fight for justice as if it were our own.
This balance of skill, experience, and empathy reflects our core philosophy that justice should not only compensate the injured, but also make healthcare safer nationwide.

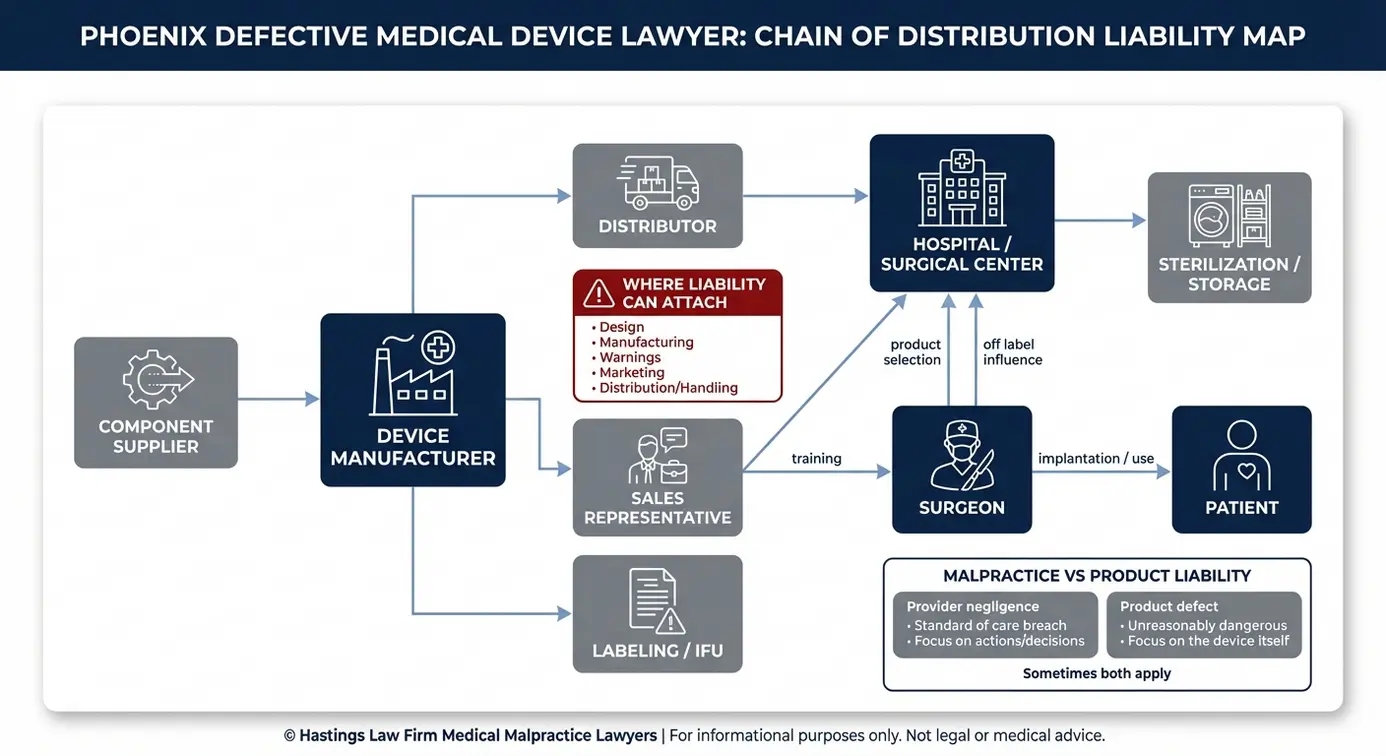
Identifying Liability: The Chain of Distribution
Liability in a defective device case often extends well beyond the manufacturer. Component suppliers, distributors, sales representatives, and in some situations hospitals that knowingly used recalled devices may all share responsibility for the harm caused through the chain of distribution. This chain refers to the various entities that handle a product from the factory to the patient.
The Manufacturer
The manufacturer is typically the primary target in a defective medical device case. If the product had a flawed design or if the company failed to warn doctors and patients about known risks, the manufacturer bears direct responsibility. A failure to warn is a marketing defect characterized by the manufacturer’s failure to adequately communicate risks through labeling or instructions despite being aware of dangers.
The Hospital or Doctor
There is an important distinction between medical malpractice and product liability. If a surgeon made an error during implantation, that may be a malpractice claim. If the device itself was defective, that is a product liability claim.
In some cases, both apply. A hospital may also face liability for hospital negligence if it continued using a device after a recall was issued or failed to follow reporting protocols. As a defective medical device lawyer team that also handles malpractice, our team includes former hospital nurses and board-certified patient advocates who help evaluate records and identify care breaches. We investigate both angles to establish causation and ensure no responsible party is overlooked.
Sales Representatives and Distributors
Distributors and sales representatives are part of the chain of distribution, meaning they can be held accountable if they contributed to the harm. Patients must show that these parties played a role in the injury. The U.S. Food and Drug Administration’s MDR Data Files track adverse event reports and can reveal whether distributors or reps were aware of problems before injuries occurred.
Role of Pharmaceutical Sales Reps in Liability
Sales representatives often have a physical presence in the operating room during implant procedures. Their role may include advising surgeons on device selection, positioning, and technique. In some cases, reps promote off-label use, which means encouraging a doctor to use a device for a purpose or in a population that the FDA has not specifically approved.
If a sales representative pushed a product for an unapproved application or failed to relay known safety warnings to the surgeon, that representative and their employer may be liable. Our team works with expert witnesses to examine communications between reps, surgeons, and manufacturers to determine whether critical safety information was withheld.
As a Phoenix product liability attorney team, we trace every link in this chain because identifying all responsible parties directly affects the strength and value of your case.
The FDA 510(k) Loophole and Federal Preemption
The FDA’s 510(k) clearance process allows medical devices to enter the market without rigorous human testing if the manufacturer can show the product is “substantially equivalent” to a device already on the market. This process often leads to unforeseen safety issues because the new device may never undergo independent clinical trials.
The 510(k) Process
510(k) clearance is a premarket review pathway that allows manufacturers to avoid the more demanding Premarket Approval (PMA) process required for higher-risk devices. This pathway primarily requires demonstrating that a device is substantially similar to an existing, legally marketed device, sometimes called a “predicate.” The problem is that the predicate device itself may have been cleared through the same pathway, creating a chain of approvals built on limited safety data.
Federal Preemption
Manufacturers sometimes argue that because the FDA cleared the device, state-level lawsuits should be blocked. This legal defense, known as federal preemption, claims that FDA oversight replaces state tort law. However, a skilled defective medical device attorney can often overcome this argument by showing that the manufacturer misled the FDA, violated the conditions of its clearance, or failed to comply with Medical Device Reporting (MDR) requirements.
You can report adverse events or review required forms through the U.S. Food and Drug Administration’s MedWatch Forms for FDA Safety Reporting.
Common Myths About FDA Clearance
- Myth: “FDA approved” means the device was proven safe in clinical trials. Fact: Most devices reach the market through 510(k) clearance, which typically does not require clinical trials.
- Myth: If the FDA cleared a device, you cannot sue the manufacturer. Fact: Federal preemption defenses can often be defeated, particularly when the manufacturer withheld safety data or violated its clearance conditions.
- Myth: The FDA actively monitors every device on the market. Fact: The FDA relies heavily on manufacturer self-reporting through MDR, and delays or underreporting of adverse events are well-documented concerns.
Mass Torts vs. Class Actions: How Your Case Is Handled
Most defective medical device cases are handled as individual lawsuits consolidated into Multi-District Litigation (MDL), not class actions. This structure ensures that each plaintiff receives compensation based on their specific injuries rather than a one-size-fits-all payout.
Why Not a Class Action?
In a class action, all members of the group typically receive the same settlement amount regardless of the severity of their injuries. That model does not work well for defective device cases because the harm varies widely from person to person. One patient may need a single revision surgery, while another may suffer permanent disability. A class action would flatten those differences and undervalue the most seriously injured individuals.
How MDL Works
MDL consolidates similar lawsuits from across the country into one federal court for pretrial proceedings, including discovery and depositions. Most defective medical device cases are handled as mass tort claims in the form of individual lawsuits to allow for this efficiency. This avoids duplicating effort across hundreds of courtrooms. However, each case retains its individual identity. If your case does not settle during the MDL process, it can be sent back to the original court for trial. You can learn more about this structure through the Federal Judicial Center’s overview of Multidistrict Litigation (MDL).
| Feature | Class Action | Multi-District Litigation (MDL) |
|---|---|---|
| How you join | Automatically included unless you opt out | File an individual lawsuit that is transferred to the MDL court |
| Compensation | Same amount for all members | Based on your individual injuries and damages |
| Case control | One representative plaintiff speaks for the group | You retain your own attorney and case strategy |
| Trial option | Rare; usually settled as a group | Individual cases can go to trial if needed |
The Hastings Approach
As a Phoenix medical device law firm, we are not a settlement mill. Even when a case is part of an MDL, we prepare every client’s claim as though it will go to trial. This means building a detailed record of your specific injuries and medical history to improve your negotiating position. This level of preparation ensures you are not treated as just another number in a mass proceeding.
Damages: Compensation for Life-Altering Complications
Patients harmed by defective medical devices may recover damages for revision surgeries, past and future medical costs, lost wages, pain and suffering, and in cases involving gross negligence, punitive damages. Recovering these costs helps ensure a family’s financial security while the patient recovers from a preventable injury.
The compensation available in a defective device case generally falls into categories of compensatory damages, which include both economic and non-economic losses, and punitive damages.
- Economic damages cover the financial losses directly tied to the injury. This includes the cost of revision surgery, which is a follow-up operation to remove, repair, or replace the failed device. It also includes rehabilitation, physical therapy, lost income, and any future medical care you are expected to need.
- Non-economic damages address the personal toll of the injury. Chronic pain, loss of mobility, emotional distress, and diminished quality of life all fall into this category. These losses do not come with a receipt, but they are real and recoverable under Arizona law.
- Punitive damages may be awarded when the evidence shows that the manufacturer acted with extreme disregard for patient safety. If a company concealed known risks or falsified safety data, punitive damages serve to hold that manufacturer accountable beyond ordinary compensation. For many of our clients, this aspect of a case is about more than money. It reflects a desire to prevent the same harm from happening to someone else.
In cases where a defective device caused a patient’s death, surviving family members may pursue a wrongful death claim to recover funeral expenses, loss of financial support, and loss of companionship.
Critical Evidence: Preserving the Explanted Device
The explanted device is the most important piece of physical proof in a defective medical device case. It must be preserved intact and kept out of the hands of the manufacturer or hospital, as it may otherwise be destroyed or misplaced.
An explant is a medical device that has been surgically removed from a patient’s body. Once removed, that device becomes the evidence of what failed and why. Without it, proving a design or manufacturing defect becomes significantly harder.
Hospitals often have standard policies to return removed devices to the manufacturer. In some cases, the device may be disposed of as biological waste. Either outcome can eliminate the evidence your case depends on. That is why legal intervention for evidence preservation before your revision surgery is so important.
What to Do Before Revision Surgery
- Request in writing that the hospital preserve the explanted device and all associated packaging, labels, and lot numbers.
- Contact a defective medical device legal team before your surgery date so your attorney can send a formal preservation letter to the hospital and manufacturer.
- Do not sign any documents authorizing the hospital to return the device to the manufacturer without consulting your attorney first.
- Ask your surgeon to photograph the device in place and document its condition at the time of removal.
- Ensure the device is stored in a sealed, labeled container and that a clear chain of custody is maintained from the operating room forward.
The U.S. District Court for the District of Arizona’s Explant Preservation Agreement in Profemur Hip Implant MDL No. 2949 provides an example of how courts formalize device preservation requirements in litigation.
Arizona Statute of Limitations for Product Liability
In Arizona, the statute of limitations for a product liability claim is generally two years from the date the injury occurred or was discovered. Missing this deadline can permanently bar your right to file a lawsuit, regardless of the strength of your case. The discovery rule, which starts the clock once you identify the cause of your injury, can be vital for patients with slowly developing complications.
The Two-Year Rule
Under Arizona Revised Statute § 12-542, you have two years to file a personal injury or product liability lawsuit. This applies to defective medical device claims as well as wrongful death actions, where the two-year clock begins on the date of the patient’s death.
The Discovery Rule
In many device cases, the injury is not immediately apparent. Metallosis, which is a condition involving metal debris in the body from a failing hip implant, for example, may develop gradually over months or years. Arizona’s discovery rule accounts for this by starting the statute of limitations clock on the date you knew, or reasonably should have known, that the device was the cause of your symptoms. This means your deadline may not begin on the date of your original implant surgery but rather on the date a doctor identified the device as the source of the problem.
Outer Time Limits
Although Arizona has a product liability statute (A.R.S. § 12-551) that originally set a twelve-year outer boundary on claims measured from the date a product was first sold, the Arizona Supreme Court declared that statute unconstitutional in *Hazine v. Montgomery Elevator Co.* (1993). As a result, Arizona does not currently enforce a product liability statute of repose. However, because the legal landscape can change, early legal consultation remains essential to protect your rights.
Do not assume you have time. If you suspect a medical device has caused you harm, contact a Phoenix defective medical device lawyer as soon as possible. Filing deadlines are strict, and preserving evidence early strengthens your case. The FDA’s Premarket Notification 510(k) page can help you research the approval history of the device in question.
Contact the Phoenix Medical Device Attorneys at Hastings Law Firm Today for Help
At Hastings Law Firm, our entire team is dedicated to medical negligence and product liability cases. Our attorneys, including former defense counsel who know how manufacturers and hospitals build their cases, work alongside in-house medical professionals to investigate every detail.
Founded by board-certified trial lawyer Tommy Hastings, our firm prepares every case as if it is going to trial because that is how we ensure your claim is taken seriously. We focus on securing full compensation for injured patients and enforcing accountability in the healthcare system.
You pay no attorney fees or costs unless we recover compensation for you. Call us today or reach out online for a free, confidential case evaluation. We can review what happened, explain your legal options, and help you take the first step toward answers and accountability.
Frequently Asked Questions About Defective Medical Device in Phoenix

Key Defective Medical Device Terms:
- Design defect
- A flaw in the blueprint or concept of a medical device that makes it unreasonably dangerous for patients, even when manufactured and used exactly as intended. In a product liability case, this means the device was unsafe before it was ever built—for example, a hip implant designed with metal-on-metal components that inevitably release toxic metal particles into the body.
- Manufacturing defect
- An error that occurs during the production process of a medical device, resulting in a specific unit or batch that differs from the intended safe design. Unlike a design defect, the blueprint was sound, but contamination, improper assembly, or quality control failures made certain devices dangerous—such as pacemakers with faulty wiring in one production run.
- Failure to warn (marketing defect)
- A type of product liability claim where a manufacturer knew or should have known about serious risks associated with a medical device but failed to provide adequate warnings or instructions to doctors and patients. This includes hiding safety data, downplaying dangers, or omitting critical information from product labels to protect sales and profits.
- FDA recall
- An action taken by a medical device manufacturer, either voluntarily or under pressure from the Food and Drug Administration, to remove or correct a product that poses a risk to public health. Recalls are classified by severity: Class I involves serious injury or death, Class II involves temporary harm, and Class III involves minor violations. A recall often serves as key evidence in a defective device lawsuit.
- Implant failure
- The malfunction, breakdown, or rejection of a surgically placed medical device inside the body, such as a hip replacement that loosens, a pacemaker that stops working, or surgical mesh that erodes through tissue. Implant failure can result from design flaws, manufacturing errors, or the body’s adverse reaction, and often requires additional surgery to remove or replace the device.
- Off-label use
- The practice of prescribing or using a medical device or drug for a purpose, patient population, or condition that the FDA has not officially approved. While doctors have discretion to use products off-label, manufacturers are prohibited from marketing devices for unapproved uses. In liability cases, off-label promotion by sales representatives can expose the manufacturer to legal responsibility if patients are harmed.
- 510(k) clearance
- A streamlined FDA approval process that allows medical device manufacturers to bring a product to market by demonstrating it is substantially similar to a device already on the market, rather than conducting rigorous safety and effectiveness testing. Critics call this a loophole because dangerous devices can enter the market based on comparison to older products that may themselves be unsafe or recalled.
- Medical Device Reporting (MDR)
- A mandatory FDA surveillance system that requires manufacturers, hospitals, and healthcare facilities to report serious injuries, malfunctions, and deaths associated with medical devices. Delays or failures in MDR reporting can indicate a manufacturer’s attempt to hide safety problems, and these records are critical evidence in defective device litigation.
- Revision surgery
- A follow-up surgical procedure required to repair, replace, or remove a failed medical device implant. Revision surgeries are often more complex, risky, and painful than the original operation, and they result in additional medical costs, recovery time, and complications. The need for revision surgery is a key component of damages in defective device cases.
- Explant
- The surgical removal of a medical device from the body, typically because the implant has failed, caused harm, or been recalled. The explanted device itself is crucial physical evidence in a product liability lawsuit, as experts can examine it for defects, wear patterns, contamination, or design flaws. Patients should ensure the device is preserved and not returned to the manufacturer without legal safeguards.
- 12-542 Injury to person injury when death ensues injury to property conversion of property forcible entry and forcible detainer two year limitation | Arizona Legislature
- Premarket Notification 510k | U.S. Food and Drug Administration
- Medical Device Recalls and Early Alerts | U.S. Food and Drug Administration
- MedWatch Forms for FDA Safety Reporting | U.S. Food and Drug Administration
- Multidistrict Litigation (MDL) | Federal Judicial Center
- MDR Data Files | U S Food and Drug Administration
- PROFEMUR HIP IMPLANT MDL NO 2949 PROD EXPLANT PRESERVATION AGREEMENT | U.S. District Court for the District of Arizona
Get Answers Today
If you think that medical negligence, a dangerous drug, or a failed medical product caused harm to you or someone you love, our team is standing by to offer guidance. We’ll explain your options under current laws and help you move forward with clarity and understanding. Case reviews are free and 100% confidential.
