

Abogado especializado en dispositivos médicos defectuosos en Phoenix
Escrito por: Bufete Hastings | Revisado por: Tommy Hastings | Actualizado: 6 de mayo de 2026
Los dispositivos médicos defectuosos pueden fallar dentro del cuerpo y provocar que los pacientes sufran complicaciones dolorosas, procedimientos adicionales y daños duraderos. La responsabilidad por productos defectuosos se centra en determinar si un dispositivo era irrazonablemente peligroso debido a un defecto de diseño, un defecto de fabricación o una falta de advertencia, y la responsabilidad puede extenderse más allá del fabricante a lo largo de la cadena de distribución. Las autorizaciones y las retiradas del mercado de la FDA pueden influir en la forma en que se evalúan estas reclamaciones, y conservar un dispositivo explantado puede ser fundamental para comprender qué falló. Si usted o un ser querido sufrieron daños o algo peor debido a un dispositivo médico defectuoso en Phoenix, Arizona, póngase en contacto con Hastings Law Firm para una revisión gratuita y confidencial de su caso.

Abogados de confianza especializados en responsabilidad civil por productos médicos en Phoenix
Lo que debe saber sobre las reclamaciones por fallos en equipos médicos en Phoenix:
- La indemnización puede depender de si el daño está relacionado con un defecto de diseño, un defecto de fabricación o la falta de advertencia.
- La responsabilidad puede extenderse más allá del fabricante cuando otras entidades de la cadena de distribución hayan contribuido al daño.
- Las opciones pueden verse limitadas cuando los fabricantes alegan la prevalencia de la legislación federal en relación con la autorización de la FDA.
- La indemnización puede incluir gastos médicos, salarios perdidos y daños morales cuando un dispositivo defectuoso provoca complicaciones que alteran la vida.
- Es posible que se puedan reclamar indemnizaciones por muerte por negligencia cuando un dispositivo defectuoso provoque la muerte de un paciente.
- Los resultados de los casos pueden variar considerablemente, ya que muchas demandas relacionadas con dispositivos médicos se tramitan como demandas individuales en litigios multidistritales, en lugar de como acciones colectivas.
- Según la legislación de Arizona, se puede perder el derecho a presentar una demanda si no se respetan los plazos de presentación.
- Demostrar la existencia de un defecto puede resultar mucho más difícil si el dispositivo explantado se devuelve al fabricante o se destruye.
- Las controversias sobre la causalidad pueden influir en el resultado cuando la lesión también podría atribuirse a una afección subyacente.
- Los datos sobre el historial de retiradas del mercado y la notificación de eventos adversos pueden ser fundamentales a la hora de evaluar si se conocían los riesgos antes de que se produjeran las lesiones.

Un bufete centrado en la atención sanitaria
Cuando un dispositivo médico falla dentro de tu cuerpo, las consecuencias pueden ser devastadoras. Confiaste en el dispositivo, en el cirujano y en el sistema que los respalda. Si esa confianza se ha traicionado, mereces respuestas y un camino claro a seguir. Como equipo de abogados especializados en dispositivos médicos defectuosos de Phoenix, Bufete Hastings se especializa exclusivamente en casos de negligencia médica y responsabilidad por productos defectuosos. Nuestro equipo legal, que cuenta con exabogados defensores y profesionales médicos internos, investiga cada eslabón de la cadena para determinar quién es responsable de su lesión. Si usted o un ser querido ha sufrido daños a causa de un implante o producto médico defectuoso, contáctenos para una consulta gratuita y confidencial evaluación de casos. Trabajamos a comisión, lo que significa que no tendrá que pagar honorarios ni gastos de abogados a menos que consigamos una indemnización para usted.
Comprender la responsabilidad por productos defectuosos: cómo los dispositivos médicos fallan a los pacientes
Una demanda por un dispositivo médico defectuoso suele basarse en uno de estos tres teorías de la responsabilidad: un defecto de diseño, un defecto de fabricación o un defecto de comercialización, lo que comúnmente se conoce como falta de advertencia. Cada teoría se centra en una etapa diferente del ciclo de vida del producto, y identificar la más adecuada es fundamental para construir un caso sólido.
La ley de responsabilidad por productos defectuosos responsabiliza a los fabricantes y a otras partes cuando un producto médico causa daños. A diferencia de una demanda por negligencia médica convencional, que se centra en las decisiones del médico, la responsabilidad por productos defectuosos se centra en el producto en sí. En virtud de la responsabilidad objetiva, se puede responsabilizar a un fabricante por las lesiones causadas por un dispositivo defectuoso, incluso sin que se demuestre negligencia o intención. Basta con que el producto sea irrazonablemente peligroso. Este estándar legal garantiza que la responsabilidad de la seguridad recaiga en el creador del producto y no en el paciente, eliminando la necesidad de demostrar que el fabricante actuó con malicia.
Estos son los tres tipos principales de defectos y cómo se aplican:
- Defecto de diseño: Un defecto de diseño es una falla presente en los planos del producto que hace que el dispositivo sea intrínsecamente inseguro incluso antes de su fabricación. Esta falla afecta a todas las unidades producidas. Los implantes de cadera «metal contra metal» son un ejemplo muy conocido; el propio diseño provocaba la liberación de partículas metálicas en el tejido circundante, lo que dio lugar a complicaciones graves en miles de pacientes.
- Defecto de fabricación: Un defecto de fabricación es un error que se produce durante el proceso de producción o montaje. Esto significa que, aunque el diseño fuera correcto, algo salió mal en una unidad o lote concreto. La contaminación durante la producción, una esterilización inadecuada o un montaje defectuoso pueden provocar defectos que hagan que un dispositivo resulte peligroso, mientras que otros de la misma línea funcionan según lo previsto.
- Defecto de comercialización (omisión de advertencia): Esta teoría se aplica cuando un fabricante tenía conocimiento de los riesgos asociados al dispositivo, pero no informó adecuadamente de dichos riesgos a los médicos o a los pacientes. Si las advertencias eran incompletas, engañosas o inexistentes, y un paciente sufrió lesiones como consecuencia de ello, el fabricante puede ser considerado responsable.
Un abogado especializado en dispositivos médicos defectuosos de Phoenix debe determinar qué teoría sobre el defecto es aplicable basándose en las pruebas. En muchos casos, hay más de una teoría relevante. Nuestros abogados especializados en dispositivos médicos defectuosos colaboran con expertos médicos para analizar el dispositivo, revisar los datos clínicos e identificar exactamente dónde se produjo el fallo.

Dispositivos médicos de alto riesgo y que suelen ser objeto de retiradas del mercado
Numerosos dispositivos se han enfrentado a Retiradas del mercado por la FDA debido a las elevadas tasas de fallos, entre los que se incluyen las prótesis de cadera metal-metal, las mallas transvaginales, los aparatos de CPAP y ciertos implantes cardíacos. Estos productos se utilizaron en miles de pacientes antes de que salieran a la luz las preocupaciones sobre su seguridad.
Un fabricante o la FDA emiten una orden de retirada para corregir un dispositivo defectuoso o para hacer frente a un riesgo de lesiones. Estas retiradas pueden ir desde correcciones menores en el etiquetado hasta retiradas completas del mercado. Puede consultar las medidas actuales y anteriores a través de la Retiradas de productos y alertas tempranas de la Administración de Alimentos y Medicamentos de EE. UU. (FDA) base de datos.
El fallo de un implante, que se refiere al mal funcionamiento o la rotura de un dispositivo dentro del cuerpo, puede provocar dolor, infección, daño tisular o la necesidad de una cirugía de revisión. En la tabla siguiente se enumeran algunas de las categorías de dispositivos más comúnmente afectadas.
| Tipo de dispositivo | Complicación frecuente | Lesiones típicas |
|---|---|---|
| Prótesis de cadera metal-metal | Metalosis, aflojamiento de componentes | Necrosis tisular, dolor crónico, cirugía de revisión |
| Malla transvaginal | Erosión a través del tejido | Infección, perforación de órganos, dolor crónico |
| Malla de hernia | Adhesión, migración de la malla | Obstrucción intestinal, reintervención |
| Aparatos de CPAP (por ejemplo, Philips Respironics) | Degradación de la espuma tóxica | Daños respiratorios, posible exposición a sustancias cancerígenas |
| Dispositivos cardíacos (marcapasos, desfibriladores) | Rotura del cable, fallo de la batería | Parada cardíaca, descarga eléctrica, cirugía de urgencia |
| Implantes de rodilla y hombro | Aflojamiento, desgaste de los componentes | Pérdida ósea, inestabilidad, cirugía de revisión |
Si ha sufrido complicaciones relacionadas con alguno de estos tipos de dispositivos, un abogado especializado en dispositivos médicos en Phoenix puede evaluar si su lesión está relacionada con un defecto conocido o una retirada del mercado. Algunos de estos productos se utilizaron fuera de lo indicado en la etiqueta, lo que significa que se implantaron para un fin no aprobado específicamente por la FDA, lo que puede reforzar aún más su demanda.
The Hastings Law Firm Diferencia
Los resultados importan, pero lo que realmente nos diferencia es cómo los conseguimos. Cada veredicto, cada acuerdo, y cada victoria en los tribunales de Phoenix viene de una promesa guía: Tratar la lucha de cada cliente por la justicia como si fuera la nuestra.
Este equilibrio de capacidad, experiencia y empatía refleja nuestra filosofía fundamental de que la justicia no sólo debe compensar a los perjudicados, sino también hacer más segura la asistencia sanitaria en todo el país.

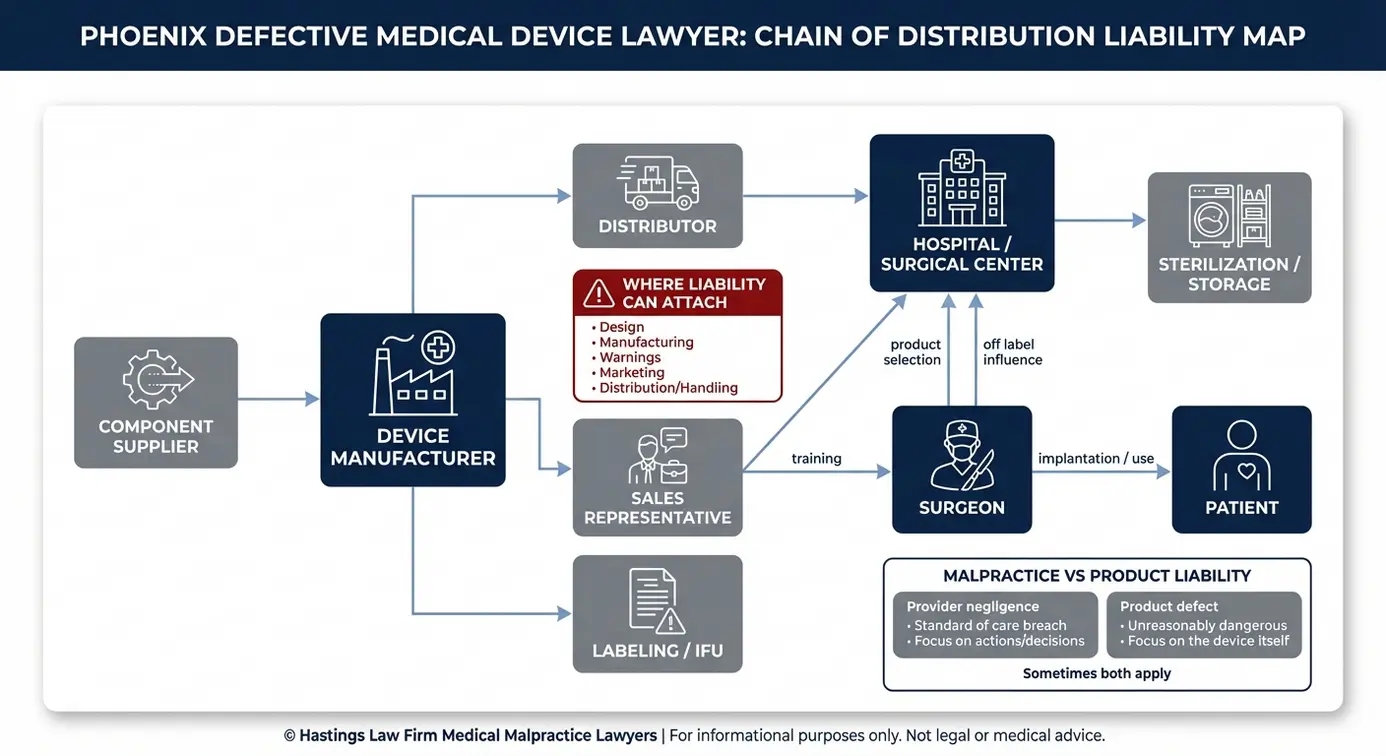
Determinación de la responsabilidad: la cadena de distribución
En los casos de dispositivos defectuosos, la responsabilidad suele ir mucho más allá del fabricante. Los proveedores de componentes, los distribuidores, los representantes de ventas y, en algunos casos, los hospitales que hayan utilizado a sabiendas dispositivos retirados del mercado pueden compartir la responsabilidad por los daños causados por el cadena de distribución. Esta cadena hace referencia a las distintas entidades que gestionan un producto desde la fábrica hasta el paciente.
El fabricante
En los casos relacionados con dispositivos médicos defectuosos, el fabricante suele ser el principal responsable. Si el producto presentaba un defecto de diseño o si la empresa no advirtió a los médicos y pacientes sobre los riesgos conocidos, el fabricante es responsable responsabilidad directa. La falta de advertencia es un defecto de comercialización que se caracteriza por el hecho de que el fabricante no comunique adecuadamente los riesgos a través del etiquetado o las instrucciones, a pesar de ser consciente de los peligros.
El hospital o el médico
Existe una distinción importante entre la negligencia médica y la responsabilidad por productos defectuosos. Si un cirujano cometió un error durante la implantación, eso podría constituir una demanda por negligencia médica. Si el dispositivo en sí era defectuoso, se trata de una demanda por responsabilidad por productos defectuosos.
En algunos casos, se aplican ambas cosas. Un hospital también puede incurrir en responsabilidad por negligencia hospitalaria si siguió utilizando un dispositivo después de que se emitiera una orden de retirada del mercado o no cumplió con los protocolos de notificación. Como equipo de abogados especializados en dispositivos médicos defectuosos que también se ocupa de casos de negligencia médica, contamos con exenfermeras de hospital y defensores de pacientes certificados por la junta profesional que ayudan a evaluar los expedientes e identificar las faltas en la atención prestada. Investigamos ambos aspectos para establecer la relación de causalidad y asegurarnos de que no se pase por alto a ninguna de las partes responsables.
Representantes de ventas y distribuidores
Los distribuidores y los representantes de ventas forman parte de la cadena de distribución, lo que significa que pueden ser considerados responsables si contribuyeron al daño. Los pacientes deben demostrar que estas partes tuvieron un papel en la lesión. El Archivos de datos MDR de la Administración de Alimentos y Medicamentos de EE.UU. pista notificaciones de reacciones adversas y puede revelar si los distribuidores o los representantes estaban al tanto de los problemas antes de que se produjeran las lesiones.
El papel de los representantes de ventas farmacéuticas en materia de responsabilidad civil
Los representantes de ventas suelen estar presentes físicamente en el quirófano durante las intervenciones de implante. Su función puede consistir en asesorar a los cirujanos sobre la selección del dispositivo, su colocación y la técnica a seguir. En algunos casos, los representantes promocionan uso no indicado en la ficha técnica, lo que significa animar a un médico a utilizar un dispositivo para un fin o en una población que la FDA no ha aprobado específicamente.
Si un representante de ventas promocionara un producto para un solicitud no aprobada o no transmitió al cirujano las advertencias de seguridad conocidas, ese representante y su empleador podrían incurrir en responsabilidad. Nuestro equipo colabora con peritos para analizar las comunicaciones entre los representantes, los cirujanos y los fabricantes, con el fin de determinar si se ocultó información de seguridad fundamental.
Como equipo de abogados especializados en responsabilidad por productos defectuosos en Phoenix, analizamos cada eslabón de esta cadena, ya que identificar a todas las partes responsables influye directamente en la solidez y el valor de su caso.
La laguna jurídica del procedimiento 510(k) de la FDA y la prevalencia federal
El proceso de autorización 510(k) de la FDA permite que los dispositivos médicos salgan al mercado sin necesidad de someterse a rigurosos ensayos en seres humanos si el fabricante puede demostrar que el producto es “sustancialmente equivalente” a un dispositivo que ya se encuentra en el mercado. Este proceso suele dar lugar a problemas de seguridad imprevistos, ya que es posible que el nuevo dispositivo nunca se someta a ensayos clínicos independientes.
El proceso 510(k)
La autorización 510(k) es una vía de revisión previa a la comercialización que permite a los fabricantes evitar los requisitos más exigentes Autorización previa a la comercialización (PMA) requerido para los dispositivos de mayor riesgo. Esta vía exige principalmente demostrar que un dispositivo es sustancialmente similar a un dispositivo ya existente y comercializado legalmente, lo que a veces se denomina “dispositivo de referencia”. El problema es que el propio dispositivo de referencia puede haber sido autorizado a través de la misma vía, lo que crea una cadena de autorizaciones basada en datos de seguridad limitados.
Prevalencia federal
Los fabricantes suelen argumentar que, dado que la FDA autorizó el dispositivo, las demandas a nivel estatal deberían ser desestimadas. Esta defensa legal, conocida como prevalencia federal, sostiene que la supervisión de la FDA sustituye a la legislación estatal en materia de responsabilidad civil. Sin embargo, un abogado especializado en casos de dispositivos médicos defectuosos suele poder rebatir este argumento demostrando que el fabricante engañó a la FDA, incumplió las condiciones de su autorización o no cumplió con los requisitos del Sistema de Notificación de Dispositivos Médicos (MDR).
Puede notificar eventos adversos o consultar los formularios necesarios a través de la Formularios MedWatch de la Administración de Alimentos y Medicamentos de EE. UU. (FDA) para la notificación de incidentes de seguridad.
Mitos comunes sobre la autorización de la FDA
- Mito: “Aprobado por la FDA” significa que se ha demostrado la seguridad del dispositivo en ensayos clínicos. Hecho: La mayoría de los dispositivos llegan al mercado tras obtener la autorización 510(k), que normalmente no exige ensayos clínicos.
- Mito: Si la FDA ha autorizado un dispositivo, no se puede demandar al fabricante. Hecho: Las defensas basadas en la prevalencia federal suelen poder refutarse, sobre todo cuando el fabricante ha ocultado datos de seguridad o ha incumplido las condiciones de autorización.
- Mito: La FDA supervisa activamente todos los dispositivos que se comercializan. Hecho: La FDA depende en gran medida de la información que los fabricantes proporcionan voluntariamente a través del MDR, y los retrasos o la falta de notificación de los eventos adversos son motivos de preocupación bien documentados.
Demandas colectivas frente a acciones colectivas: cómo se gestiona tu caso
La mayoría de los casos relacionados con dispositivos médicos defectuosos se tramitan como demandas individuales que se agrupan en Litigios multidistritales (MDL), y no demandas colectivas. Esta estructura garantiza que cada demandante reciba una indemnización basada en los daños específicos que haya sufrido, en lugar de un pago único para todos.
¿Por qué no una demanda colectiva?
En una demanda colectiva, todos los miembros del grupo suelen recibir lo mismo importe de la liquidación independientemente de la gravedad de sus lesiones. Ese modelo no funciona bien en los casos de dispositivos defectuosos, ya que el daño varía mucho de una persona a otra. Un paciente puede necesitar una sola cirugía de revisión, mientras que otro puede sufrir una discapacidad permanente. Una demanda colectiva nivelaría esas diferencias y subestimaría el valor de las personas más gravemente lesionadas.
Cómo funciona MDL
El MDL agrupa demandas similares de todo el país en un solo tribunal federal para procedimientos previos al juicio, incluyendo la fase de presentación de pruebas y las declaraciones. La mayoría de los casos relacionados con dispositivos médicos defectuosos se tramitan como demandas colectivas en forma de acciones judiciales individuales para permitir esta eficiencia. De este modo se evita la duplicación de esfuerzos en cientos de tribunales. Sin embargo, cada caso conserva su identidad individual. Si su caso no se resuelve mediante un acuerdo durante el proceso de MDL, puede remitirse de nuevo al tribunal original para que se celebre un juicio. Puede obtener más información sobre esta estructura a través de la Resumen del Centro Judicial Federal sobre los litigios multidistritales (MDL).
| Característica | Demanda colectiva | Litigio multidistrital (MDL) |
|---|---|---|
| Cómo participar | Se incluye automáticamente a menos que se opte por no participar | Interponer una demanda individual que se remita al tribunal del MDL |
| Compensación | La misma cantidad para todos los miembros | En función de sus lesiones y daños específicos |
| Estudio de casos y controles | Un demandante representativo actúa en nombre del grupo | Usted contrata a su propio abogado y decide la estrategia del caso |
| Versión de prueba | Poco frecuente; suele vivir en grupo | Los casos individuales pueden llegar a juicio si es necesario |
El enfoque de Hastings
Como bufete de abogados especializado en dispositivos médicos con sede en Phoenix, no somos una fábrica de acuerdos extrajudiciales. Incluso cuando un caso forma parte de un MDL, preparamos la demanda de cada cliente como si fuera a llegar a juicio. Esto implica elaborar un expediente detallado de sus lesiones específicas y su historial médico para mejorar su posición negociadora. Este nivel de preparación garantiza que no te traten como un simple número más en un proceso masivo.
Indemnización por daños y perjuicios: compensación por complicaciones que alteran la vida
Los pacientes que hayan sufrido daños a causa de dispositivos médicos defectuosos pueden reclamar una indemnización por los gastos de las cirugías de revisión, los gastos médicos pasados y futuros, los salarios perdidos, el dolor y el sufrimiento, y, en los casos de negligencia grave, una indemnización punitiva. La recuperación de estos gastos contribuye a garantizar la seguridad económica de la familia mientras el paciente se recupera de una lesión que se podría haber evitado.
Las indemnizaciones que pueden obtenerse en un caso por un dispositivo defectuoso suelen clasificarse en las siguientes categorías: daños compensatorios, que incluyen tanto pérdidas económicas como no económicas, así como indemnizaciones punitivas.
- Daños económicos cubrir las pérdidas económicas directamente relacionadas con la lesión. Esto incluye el costo de la cirugía de revisión, que es una operación de seguimiento para retirar, reparar o reemplazar el implante defectuoso. También incluye la rehabilitación, la fisioterapia, la pérdida de ingresos y cualquier atención médica futura que se prevea que necesites.
- Daños no económicos abordar las consecuencias personales de la lesión. El dolor crónico, la pérdida de movilidad, el sufrimiento emocional y la disminución de la calidad de vida entran todos en esta categoría. Estas pérdidas no vienen acompañadas de un recibo, pero son reales y se pueden reclamar según la legislación de Arizona.
- Daños punitivos pueden concederse cuando las pruebas demuestran que el fabricante actuó con un desprecio extremo por la seguridad de los pacientes. Si una empresa ocultó riesgos conocidos o falsificó datos de seguridad, los daños punitivos sirven para exigir responsabilidades al fabricante más allá de la indemnización ordinaria. Para muchos de nuestros clientes, este aspecto del caso va más allá del dinero. Refleja el deseo de evitar que el mismo daño le ocurra a otra persona.
En los casos en que un dispositivo defectuoso haya causado la muerte de un paciente, los familiares sobrevivientes pueden presentar una demanda por homicidio culposo para reclamar el reembolso de los gastos funerarios, la pérdida de manutención económica y la pérdida de compañía.
Pruebas fundamentales: conservación del dispositivo extraído
El dispositivo explantado es la pieza más importante de prueba física en un caso de dispositivo médico defectuoso. Debe conservarse intacto y mantenerse fuera del alcance del fabricante o del hospital, ya que, de lo contrario, podría ser destruido o extraviarse.
Un implante extraído es un dispositivo médico que se ha retirado quirúrgicamente del cuerpo de un paciente. Una vez retirado, ese dispositivo se convierte en la prueba de qué falló y por qué. Sin él, demostrar que existe un defecto de diseño o de fabricación resulta mucho más difícil.
Los hospitales suelen tener políticas estándar para devolver los dispositivos extraídos al fabricante. En algunos casos, el dispositivo puede eliminarse como residuo biológico. Cualquiera de estas dos opciones puede destruir las pruebas de las que depende su caso. Por eso es tan importante la intervención legal para la preservación de las pruebas antes de su cirugía de revisión.
Qué hacer antes de una cirugía de revisión
- Solicite por escrito al hospital que conserve el dispositivo explantado y todo el embalaje, las etiquetas y los números de lote correspondientes.
- Póngase en contacto con un equipo de abogados especializados en dispositivos médicos defectuosos antes de la fecha de su cirugía para que su abogado pueda enviar una notificación formal carta de conservación al hospital y al fabricante.
- No firme ningún documento que autorice al hospital a devolver el dispositivo al fabricante sin consultar primero con su abogado.
- Pídale a su cirujano que tome una fotografía del dispositivo colocado y que documente su estado en el momento de la extracción.
- Asegúrese de que el dispositivo se guarde en un recipiente sellado y etiquetado, y de que se mantenga una cadena de custodia clara desde el quirófano en adelante.
En Acuerdo de conservación de implantes del Tribunal de Distrito de los Estados Unidos para el Distrito de Arizona en el caso Profemur Hip Implant MDL n.º 2949 ofrece un ejemplo de cómo los tribunales establecen los requisitos de conservación de dispositivos en los litigios.
Plazo de prescripción de Arizona en materia de responsabilidad por productos defectuosos
En Arizona, el plazo de prescripción para una demanda por responsabilidad civil por productos defectuosos suele ser de dos años a partir de la fecha en que se produjo o se descubrió la lesión. Si no se respeta este plazo, se puede perder para siempre el derecho a presentar una demanda, independientemente de la solidez de su caso. El regla de descubrimiento, que inicia el conteo una vez que se identifica la causa de la lesión, puede ser vital para los pacientes con complicaciones de evolución lenta.
La regla de los dos años
En Estatuto Revisado de Arizona, § 12-542, tienes dos años para presentar una daños personales o una demanda por responsabilidad civil por productos defectuosos. Esto se aplica tanto a las reclamaciones por dispositivos médicos defectuosos como a las demandas por homicidio culposo, en las que el plazo de dos años comienza a contar a partir de la fecha del fallecimiento del paciente.
La regla del descubrimiento
En muchos casos relacionados con dispositivos, la lesión no se nota de inmediato. Metalosis, que es una afección provocada por la presencia de residuos metálicos en el cuerpo debido, por ejemplo, a un implante de cadera defectuoso, puede desarrollarse gradualmente a lo largo de meses o años. La «regla del descubrimiento» de Arizona tiene esto en cuenta al fijar el inicio del plazo de prescripción en la fecha en que usted supo, o razonablemente debería haber sabido, que el dispositivo era la causa de sus síntomas. Esto significa que es posible que su plazo no comience en la fecha de la cirugía de implante original, sino en la fecha en que un médico identificó el dispositivo como la causa del problema.
Plazos externos
Aunque Arizona cuenta con una ley de responsabilidad por productos defectuosos (A.R.S. § 12-551) que originalmente establecía un plazo máximo de doce años para presentar reclamaciones, contado a partir de la fecha en que se vendió el producto por primera vez, la Corte Suprema de Arizona declaró inconstitucional dicha ley en el caso *Hazine v. Montgomery Elevator Co.* (1993). Como resultado, Arizona no aplica actualmente un plazo de prescripción para la responsabilidad por productos defectuosos. Sin embargo, dado que el panorama legal puede cambiar, la consulta legal temprana sigue siendo esencial para proteger sus derechos.
No des por sentado que tienes tiempo. Si sospechas que un dispositivo médico te ha causado daños, ponte en contacto con un abogado especializado en dispositivos médicos defectuosos de Phoenix lo antes posible. Los plazos para presentar la demanda son estrictos, y conservar las pruebas desde el principio refuerza tu caso. El Notificación previa a la comercialización 510(k) de la FDA Esta página puede ayudarte a consultar el historial de aprobación del dispositivo en cuestión.
Póngase en contacto hoy mismo con los abogados especializados en dispositivos médicos de Phoenix, en Hastings Law Firm, para obtener ayuda
En Hastings Law Firm, todo nuestro equipo se dedica a casos de negligencia médica y responsabilidad por productos defectuosos. Nuestros abogados, entre los que se encuentran antiguos abogados defensores que conocen a la perfección cómo los fabricantes y los hospitales preparan sus argumentos, trabajan codo a codo con profesionales médicos de nuestra propia plantilla para investigar cada detalle.
Fundado por el abogado litigante certificado Tommy Hastings, nuestro bufete prepara cada caso como si fuera a llegar a juicio, ya que así es como nos aseguramos de que su reclamación se tome en serio. Nos enfocamos en conseguir una indemnización completa para los pacientes lesionados y en exigir responsabilidades al sistema de salud.
No tendrá que pagar honorarios ni gastos de abogados a menos que consigamos una indemnización para usted. Llámenos hoy mismo o póngase en contacto con nosotros a través de nuestra página web para evaluación gratuita y confidencial de su caso. Podemos analizar lo que ocurrió, explicarle sus opciones legales y ayudarle a dar el primer paso para obtener respuestas y que se rindan cuentas.
Preguntas frecuentes sobre dispositivos médicos defectuosos en Phoenix

Términos clave sobre productos sanitarios defectuosos:
- Defecto de diseño
- Un defecto en el diseño o el concepto de un dispositivo médico que lo convierte en irrazonablemente peligroso para los pacientes, incluso cuando se fabrica y utiliza exactamente según lo previsto. En un caso de responsabilidad por productos defectuosos, esto significa que el dispositivo ya era inseguro antes incluso de su fabricación; por ejemplo, un implante de cadera diseñado con componentes de metal contra metal que, inevitablemente, liberan partículas metálicas tóxicas en el cuerpo.
- Defecto de fabricación
- Un error que se produce durante el proceso de fabricación de un dispositivo médico y que da lugar a que una unidad o un lote concreto difiera del diseño seguro previsto. A diferencia de un defecto de diseño, el proyecto era correcto, pero la contaminación, un montaje inadecuado o fallos en el control de calidad hicieron que ciertos dispositivos resultaran peligrosos, como los marcapasos con cableado defectuoso en una tanda de producción.
- Omisión de advertencias (defecto de comercialización)
- Tipo de demanda por responsabilidad civil por productos defectuosos en la que el fabricante conocía o debería haber conocido los graves riesgos asociados a un dispositivo médico, pero no proporcionó las advertencias o instrucciones adecuadas a los médicos y pacientes. Esto incluye ocultar datos de seguridad, restar importancia a los peligros u omitir información fundamental en las etiquetas del producto con el fin de proteger las ventas y los beneficios.
- Retirada del mercado por la FDA
- Medida adoptada por un fabricante de dispositivos médicos, ya sea de forma voluntaria o bajo presión de la Administración de Alimentos y Medicamentos (FDA), para retirar o corregir un producto que supone un riesgo para la salud pública. Las retiradas se clasifican según su gravedad: la Clase I implica lesiones graves o la muerte, la Clase II implica daños temporales y la Clase III implica infracciones menores. Una retirada suele servir como prueba clave en una demanda por un dispositivo defectuoso.
- Fallo del implante
- El mal funcionamiento, la avería o el rechazo de un dispositivo médico implantado quirúrgicamente en el cuerpo, como una prótesis de cadera que se afloja, un marcapasos que deja de funcionar o una malla quirúrgica que se erosiona a través del tejido. El fallo de un implante puede deberse a defectos de diseño, errores de fabricación o una reacción adversa del organismo, y a menudo requiere una nueva intervención quirúrgica para retirar o sustituir el dispositivo.
- Uso no indicado en la ficha técnica
- La práctica de recetar o utilizar un dispositivo médico o un medicamento para un fin, una población de pacientes o una afección que la FDA no ha aprobado oficialmente. Si bien los médicos tienen la facultad de utilizar productos fuera de lo indicado en la etiqueta, los fabricantes tienen prohibido comercializar dispositivos para usos no aprobados. En casos de responsabilidad civil, la promoción fuera de lo indicado en la etiqueta por parte de los representantes de ventas puede exponer al fabricante a responsabilidades legales si los pacientes sufren daños.
- Autorización 510(k)
- Un proceso simplificado de aprobación de la FDA que permite a los fabricantes de dispositivos médicos comercializar un producto demostrando que es sustancialmente similar a un dispositivo que ya se encuentra en el mercado, en lugar de realizar rigurosas pruebas de seguridad y eficacia. Los críticos consideran que esto es una laguna jurídica, ya que los dispositivos peligrosos pueden entrar en el mercado basándose en la comparación con productos más antiguos que, a su vez, pueden ser inseguros o haber sido retirados del mercado.
- Notificación de productos sanitarios (MDR)
- Un sistema de vigilancia obligatorio de la FDA que exige a los fabricantes, hospitales y centros de salud que notifiquen las lesiones graves, los fallos de funcionamiento y las muertes relacionadas con los dispositivos médicos. Los retrasos o incumplimientos en la presentación de informes al MDR pueden indicar un intento por parte del fabricante de ocultar problemas de seguridad, y estos registros constituyen pruebas fundamentales en los litigios por dispositivos defectuosos.
- Cirugía de revisión
- Una intervención quirúrgica de revisión necesaria para reparar, sustituir o extraer un implante médico defectuoso. Las cirugías de revisión suelen ser más complejas, arriesgadas y dolorosas que la operación original, y conllevan gastos médicos adicionales, un tiempo de recuperación más prolongado y mayores complicaciones. La necesidad de una cirugía de revisión es un elemento clave a la hora de calcular los daños y perjuicios en los casos de dispositivos defectuosos.
- Extracción
- La extracción quirúrgica de un dispositivo médico del cuerpo, normalmente debido a que el implante ha fallado, ha causado daños o ha sido objeto de una retirada del mercado. El dispositivo explantado constituye en sí mismo una prueba física crucial en un juicio por responsabilidad civil por productos defectuosos, ya que los expertos pueden examinarlo en busca de defectos, patrones de desgaste, contaminación o fallos de diseño. Los pacientes deben asegurarse de que el dispositivo se conserve y no se devuelva al fabricante sin las garantías legales pertinentes.
- 12-542 Lesión a la persona lesión cuando la muerte sobreviene lesión a la propiedad conversión de la propiedad de entrada forzosa y retención forzosa de dos años de limitación | 12-542 Lesión a la persona lesión cuando la muerte sobreviene Legislatura de Arizona
- Notificación previa a la comercialización 510(k) | Administración de Alimentos y Medicamentos de EE.UU.
- Retiradas de productos sanitarios y alertas tempranas | Administración de Alimentos y Medicamentos de EE.UU.
- Formularios MedWatch para informes de seguridad de la FDA | Administración de Alimentos y Medicamentos de EE.UU.
- Litigios multidistritales (MDL) | Centro Judicial Federal
- Archivos de datos MDR | Administración de Alimentos y Medicamentos de los Estados Unidos
- ACUERDO DE CONSERVACIÓN DE IMPLANTES DE CADERA PROFEMUR, N.º DE MODELO 2949 | Tribunal Federal de Distrito para el Distrito de Arizona
Obtenga respuestas hoy mismo
Si cree que una negligencia médica, un medicamento peligroso o un producto médico defectuoso le han causado daños a usted o a un ser querido, nuestro equipo está a su disposición para ofrecerle orientación. Le explicaremos las opciones que le ofrece la legislación vigente y le ayudaremos a avanzar con claridad y comprensión. Las revisiones de casos son gratuitas y 100% confidenciales.
