

Abogado especializado en dispositivos médicos defectuosos en Arizona
Escrito por: Bufete Hastings | Revisado por: Tommy Hastings | Actualizado: 6 de mayo de 2026
Un dispositivo médico defectuoso puede convertir un tratamiento de confianza en un dolor constante, procedimientos repetidos y trastornos duraderos. La ley de responsabilidad por productos defectuosos de Arizona se centra en determinar si un dispositivo era irrazonablemente peligroso debido a su diseño, fabricación o advertencias, y la responsabilidad objetiva puede aplicarse incluso sin necesidad de demostrar una intención negligente. Las reclamaciones también pueden solaparse con la negligencia médica cuando un error del proveedor contribuye al daño. El historial regulatorio, las retiradas del mercado y los datos de notificaciones pueden influir en la forma en que se evalúa la responsabilidad. Si usted o un ser querido sufrieron daños o algo peor debido a un dispositivo médico defectuoso en Phoenix, Arizona, póngase en contacto con Hastings Law Firm para una revisión gratuita y confidencial de su caso.

Los mejores abogados especializados en responsabilidad civil por productos médicos en Arizona
Lo que debe saber sobre las reclamaciones por fallos en equipos médicos en Arizona:
- Una falla en el dispositivo puede provocar daños que alteren la vida, como dolor, cirugías adicionales y complicaciones a largo plazo.
- Las opciones de indemnización pueden depender de si el daño está relacionado con un defecto del dispositivo o con un error del proveedor de servicios de salud, ya que la responsabilidad por productos defectuosos y la negligencia médica pueden solaparse.
- La responsabilidad puede extenderse más allá del fabricante a otros eslabones de la cadena de distribución, lo que puede influir en quién pueda ser considerado responsable.
- Demostrar que existe una relación directa entre el defecto y la lesión puede ser fundamental, ya que los fabricantes podrían argumentar que el daño se debió a una afección preexistente o a la técnica quirúrgica.
- La indemnización puede incluir gastos médicos, pérdida de ingresos y daños morales, y en casos de negligencia grave se pueden conceder daños punitivos.
- Un desenlace fatal puede hacer que la demanda se centre en una indemnización por muerte por negligencia, como los gastos del funeral y la pérdida de compañía.
- La autorización de la FDA no impide necesariamente que se presente una demanda, y la vía 510(k) puede ser relevante para determinar cómo llegó un dispositivo al mercado.
- Los datos sobre retiradas de productos y notificaciones de dispositivos médicos pueden ser importantes, ya que pueden revelar patrones de fallo y el momento en que se detectaron los riesgos.
- En Arizona, las opciones pueden verse limitadas por normas relacionadas con los plazos, ya que las demandas por responsabilidad civil por productos defectuosos suelen estar sujetas a un plazo de prescripción.
- El dispositivo extraído puede constituir una prueba fundamental, ya que su conservación determina si el dispositivo podrá examinarse posteriormente.

Un bufete centrado en la atención sanitaria
Cuando un dispositivo médico falla dentro de tu cuerpo, la experiencia puede hacerte sentir aislado. Confiabas en que el implante, el filtro o la malla eran seguros, y ahora tienes que lidiar con el dolor, nuevas cirugías y preguntas que nadie parece dispuesto a responder. Si tú o un ser querido habéis sufrido daños a causa de un producto médico defectuoso, merecéis respuestas claras y abogados con experiencia asesoramiento jurídico.
Hastings Law Firm se especializa exclusivamente en casos de negligencia médica y responsabilidad civil por productos médicos. Nuestro equipo, que cuenta con enfermeras consultoras internas y exabogados defensores, sabe cómo investigar los fallos de los dispositivos tanto desde el punto de vista médico como jurídico. Como bufete especializado en casos de dispositivos médicos defectuosos en Arizona, preparamos cada caso como si fuera a ir a juicio, porque ese nivel de preparación es lo que garantiza resultados justos.
Si cree que un dispositivo médico le causó una lesión, estaremos encantados de analizar lo sucedido y explicarle sus opciones durante una evaluación gratuita y confidencial.
Definición de productos médicos defectuosos según la legislación de Arizona
Un dispositivo médico defectuoso es cualquier instrumento médico o implante que presente un riesgo irrazonable de causar daño debido a errores en su diseño, en su fabricación o en las advertencias que lo acompañan. Según la ley de responsabilidad por productos defectuosos de Arizona, el criterio no es si el dispositivo causó una complicación, sino si el dispositivo en sí mismo presentaba un defecto que lo hiciera irrazonablemente peligroso.
Esta distinción es importante. Todo implante quirúrgico conlleva riesgos conocidos, y se espera que los médicos informen sobre dichos riesgos antes de la intervención. Una complicación conocida, como una inflamación leve tras una artroplastia, es diferente de un defecto. Se habla de defecto cuando el producto falla de una manera que va más allá de esos riesgos aceptados. Por ejemplo, un implante de cadera podría liberar partículas metálicas tóxicas en el tejido circundante debido a un diseño defectuoso del cojinete.
La negligencia médica se refiere a un error cometido por un profesional de la salud, como por ejemplo, un cirujano que coloca un implante de forma incorrecta. La responsabilidad por productos defectuosos, por otro lado, se centra en el dispositivo en sí y en las empresas responsables de su comercialización. A menudo, ambas reclamaciones coexisten. Hastings Law Firm gestiona simultáneamente las reclamaciones por negligencia médica y por responsabilidad por productos defectuosos para investigar todas las posibles causas del daño.
Se aplica a Arizona responsabilidad objetiva en los casos de responsabilidad por productos defectuosos. Esto significa que el enfoque jurídico se centra en si el dispositivo era defectuoso, y no en la intención del fabricante. Un paciente lesionado no tiene que demostrar que el fabricante actuó con negligencia. Solo tiene que demostrar que el dispositivo era defectuoso y que ese defecto causó su lesión.
Otros conceptos habituales incluyen la Notificación de Dispositivos Médicos (MDR). Se trata del sistema que utilizan los fabricantes y los centros de salud para notificar a la FDA las lesiones y los fallos de funcionamiento relacionados con los dispositivos. La retirada de un dispositivo médico, una medida formal que se adopta cuando se determina que un dispositivo presenta un riesgo de daño grave, también puede respaldar una demanda al confirmar que el producto era peligroso.

Tres tipos de defectos en dispositivos que pueden dar lugar a acciones legales
Las demandas por responsabilidad civil por productos defectuosos suelen clasificarse en tres categorías: defectos de diseño inherentes al propio producto, defectos de fabricación que se producen durante el montaje o la producción, y defectos de comercialización relacionados con la falta de advertencia a médicos o pacientes sobre riesgos conocidos. Reclamaciones de responsabilidad por productos defectuosos requieren una investigación exhaustiva del historial y el rendimiento del dispositivo.
Cada tipo de defecto requiere un enfoque diferente en cuanto a la investigación y la demostración.
Defectos de diseño
Se habla de defectos de diseño cuando toda la línea de productos es intrínsecamente peligroso, incluso cuando todas las unidades se fabrican exactamente según lo previsto. Un defecto de diseño es una falla en el plano original o en el concepto de ingeniería del producto. Esto significa que ninguna versión del dispositivo es segura. Los implantes de cadera de metal contra metal son un ejemplo muy conocido.
El propio diseño provocaba que se desprendieran residuos metálicos en el tejido de los pacientes, lo que causaba dolor crónico, necrosis tisular y la necesidad de una cirugía de revisión. Dado que el defecto se encuentra en el diseño, todos los pacientes a los que se les implantó este modelo corren riesgo, independientemente de la calidad con la que se haya realizado la cirugía.
Defectos de fabricación
Los defectos de fabricación se producen cuando un lote o una unidad concretos se contaminan, se ensamblan incorrectamente o se dañan durante la producción. Estos defectos son limitado a números de lote específicos, lo que significa que un paciente puede recibir un dispositivo que funciona correctamente, mientras que otro recibe uno peligroso de la misma línea de productos.
Puede que el diseño fuera correcto, pero algo salió mal en la planta de producción. Esto podría incluir un lote contaminado de tornillos para la columna vertebral o un cable de marcapasos con una soldadura defectuosa.
Deficiencias de marketing
Defectos de comercialización, a menudo denominados deficiencias de marketing o la falta de advertencia, surgen cuando el fabricante tenía conocimiento de los riesgos pero no los comunicó adecuadamente a los médicos o a los pacientes. Estas demandas se centran en la información y las advertencias que acompañan a un producto médico. La «falta de advertencia» es el término jurídico que designa la obligación del fabricante de comunicar los riesgos conocidos asociados a su producto.
Si los datos de los ensayos clínicos revelaran una alta tasa de fracaso y la empresa no divulgara esa información, un abogado podría fundamentar una demanda en esa falta de transparencia. Esto suele implicar demostrar que no se compartieron datos críticos de seguridad con la comunidad médica.
En Archivos de datos del MDR de la Administración de Alimentos y Medicamentos de EE. UU. son un recurso útil en estas investigaciones. Los informes de MDR presentados por hospitales, fabricantes y pacientes suelen revelar patrones de fallo que respaldan las demandas contra un dispositivo específico. Un abogado especializado en dispositivos médicos defectuosos de Arizona revisará estos informes para preparar su caso. Al analizar el momento y el volumen de estos informes, podemos determinar exactamente cuándo un fabricante tuvo conocimiento de un problema.
The Hastings Law Firm Diferencia
Los resultados importan, pero lo que realmente nos diferencia es cómo los conseguimos. Cada veredicto, cada acuerdo, y cada victoria en los tribunales de Arizona viene de una promesa guía: Tratar la lucha de cada cliente por la justicia como si fuera la nuestra.
Este equilibrio de capacidad, experiencia y empatía refleja nuestra filosofía fundamental de que la justicia no sólo debe compensar a los perjudicados, sino también hacer más segura la asistencia sanitaria en todo el país.

Dispositivos de alto riesgo que suelen ser objeto de litigios en los tribunales de Arizona
Los tribunales suelen tramitar demandas relacionadas con prótesis articulares defectuosas, mallas quirúrgicas y filtros vasculares que se desplazan o se rompen dentro del paciente. Estas categorías representan algunas de las las causas más comunes de litigios relacionados con dispositivos médicos en Arizona y en todo el país.
Los implantes ortopédicos, en particular las prótesis de cadera y rodilla, generan un volumen considerable de reclamaciones. Los implantes de cadera «metal contra metal» se han relacionado con la metalosis, un tipo de intoxicación por metales causada por la liberación de partículas en el organismo. Estos problemas pueden provocar la necrosis de los tejidos y el fallo prematuro del dispositivo, lo que requiere una cirugía de revisión, es decir, una segunda intervención para retirar y sustituir el implante original. Algunos de estos dispositivos fueron objeto de una retirada del mercado tras años de complicaciones reportadas.
Los productos de malla quirúrgica, incluidas las mallas para hernias y las mallas transvaginales, también han sido objeto de numerosos litigios. Estos productos pueden encogerse, erosionar los tejidos o provocar infecciones crónicas. Las mallas transvaginales, en particular, han dado lugar a miles de demandas individuales después de que las pacientes sufrieran perforaciones de órganos y dolores debilitantes.
Los dispositivos cardiovasculares completan la categoría que más litigios genera. Un filtro de la vena cava inferior (VCI), un pequeño dispositivo metálico que se implanta en una vena principal para atrapar coágulos sanguíneos, puede romperse, inclinarse o desplazarse hacia el corazón o los pulmones. Los electrodos defectuosos de marcapasos y desfibriladores también han sido objeto de demandas individuales y litigios a gran escala. La extracción de un electrodo roto es un procedimiento complejo que requiere habilidades quirúrgicas especializadas.
En Notificación de dispositivos médicos (MDR) de la Administración de Alimentos y Medicamentos de EE. UU. El programa registra los eventos adversos notificados para todos estos tipos de dispositivos y puede aportar pruebas importantes en una reclamación.
| Categoría de dispositivo | Modos de fallo habituales | Lesión asociada |
|---|---|---|
| Implantes de cadera y rodilla | Desprendimiento y aflojamiento de residuos metálicos | Metalosis, pérdida ósea, cirugía de revisión |
| Malla para hernias | Contracción, adhesión, migración | Dolor crónico, infección, obstrucción intestinal |
| Malla transvaginal | Erosión a través del tejido | Perforación de un órgano, dolor crónico |
| Filtros IVC | Fractura, inclinación, migración | Embolia, daño orgánico, perforación vascular |
| Electrodos para marcapasos y desfibriladores | Fractura, fallo del aislamiento | Episodios cardíacos, descargas innecesarias, cirugía de revisión |
La laguna jurídica del procedimiento 510(k) de la FDA y la responsabilidad del fabricante
Muchos dispositivos defectuosos llegan al mercado a través del proceso 510(k) de la FDA, que permite a los fabricantes eludir rigurosas pruebas de seguridad alegando que el nuevo dispositivo es “sustancialmente equivalente” a un producto anterior ya autorizado. Esta vía es uno de los aspectos más malinterpretados de la regulación de los dispositivos médicos y afecta directamente a cómo se determina la responsabilidad. El proceso 510(k) permite que los productos lleguen al mercado, a menudo sin necesidad de nuevos ensayos clínicos.
La autorización 510(k) de la FDA es una vía regulatoria que permite que un dispositivo llegue al mercado, a menudo sin necesidad de nuevos ensayos clínicos. El fabricante solo debe demostrar que el dispositivo es sustancialmente similar a otro dispositivo comercializado legalmente, conocido como «dispositivo de referencia». Esto difiere fundamentalmente de la aprobación previa a la comercialización (PMA). La PMA es el proceso de revisión más riguroso de la FDA y exige datos de ensayos clínicos que demuestren que el dispositivo es seguro y eficaz.
El problema es que la autorización 510(k) puede perpetuar defectos heredados. Si el dispositivo de referencia original tenía un diseño defectuoso, todos los dispositivos posteriores autorizados basándose en ese dispositivo de referencia pueden heredar el mismo defecto. El Guía de la Administración de Alimentos y Medicamentos de EE. UU. sobre cómo estudiar y comercializar su dispositivo describe estos procedimientos, pero el proceso regulatorio no garantiza que un dispositivo autorizado sea seguro para los pacientes.
La autorización de la FDA no equivale a una aprobación de la FDA, y no exime a los fabricantes de posibles demandas. Un abogado especializado en dispositivos médicos defectuosos de Phoenix puede explicarle cómo influye en su caso el historial regulatorio de un dispositivo específico y si el procedimiento 510(k) creó las condiciones que dieron lugar al fallo que usted sufrió.
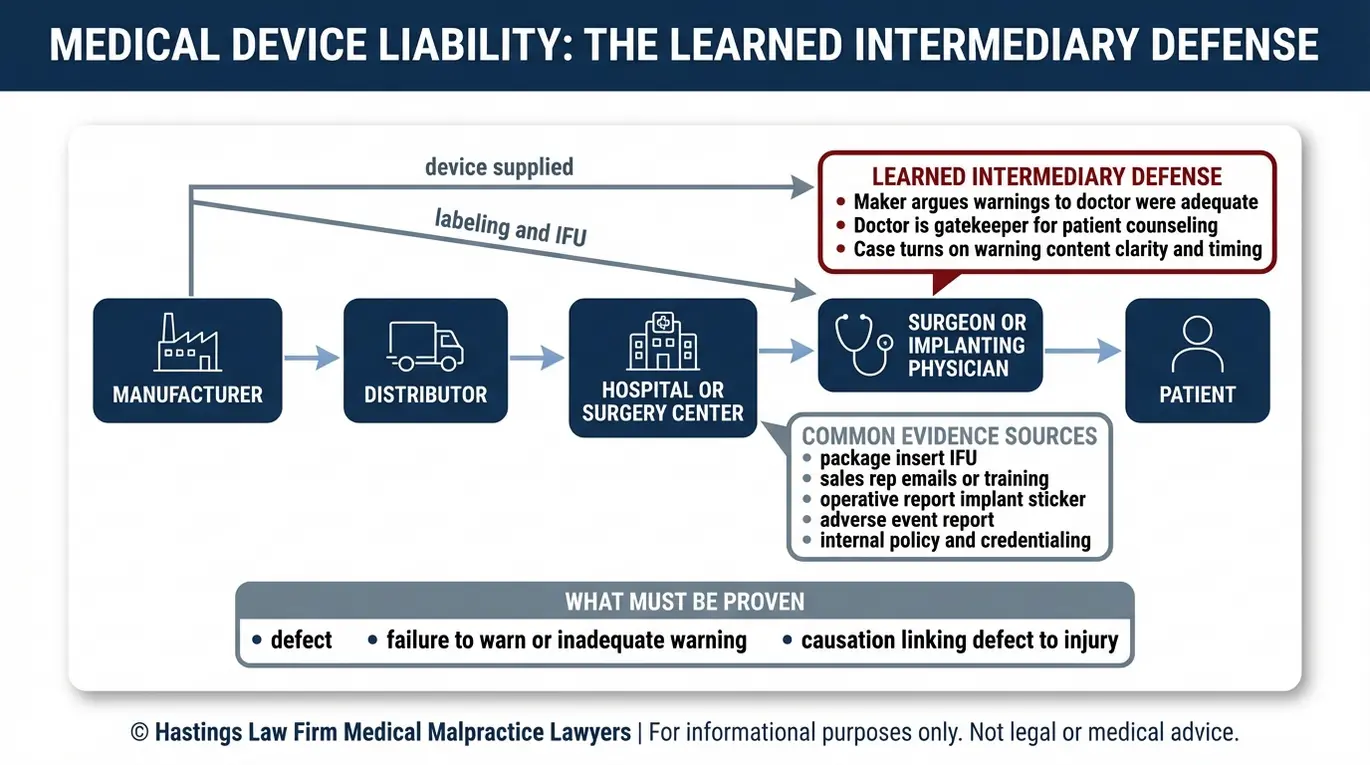
Cómo demostrar la responsabilidad y refutar la defensa del intermediario informado
Los fabricantes suelen intentar culpar al cirujano que realiza la implantación recurriendo a la doctrina del “intermediario informado”, alegando que proporcionaron las advertencias adecuadas al médico y que este, como profesional capacitado, era responsable de comunicar dichos riesgos al paciente. La responsabilidad civil se refiere a la responsabilidad legal que tiene una empresa por las lesiones que causa su producto.
La responsabilidad en un caso de dispositivo defectuoso puede extenderse a lo largo de toda la cadena de distribución. Esto incluye al fabricante que diseñó y fabricó el dispositivo, al distribuidor que lo suministró y, en ocasiones, al representante de ventas que lo promocionó ante el cirujano. En algunos casos, los hospitales o los proveedores minoristas también pueden compartir la responsabilidad si continuaron suministrando un producto del que se sabía que había sido retirado del mercado o que era defectuoso. Identificar a todas las partes responsables es una parte importante de la investigación.
En Doctrina del intermediario informado intenta romper el vínculo entre el fabricante y el paciente al introducir al médico como intermediario. Sin embargo, esta defensa no es válida cuando las advertencias proporcionadas al médico eran en sí mismas inadecuadas, engañosas o incompletas. Si un fabricante minimizó las tasas de fallo en sus materiales de comercialización o no actualizó las advertencias tras la aparición de nuevos datos sobre eventos adversos, la doctrina no lo protege.
Demostrar la causalidad, es decir, el vínculo directo entre el defecto del dispositivo y la lesión del paciente, es el elemento final y más complejo. Un abogado especializado en responsabilidad civil por productos médicos en Arizona debe demostrar que fue el defecto, y no la afección subyacente del paciente ni la técnica del cirujano, lo que causó el daño. Nuestro equipo colabora con expertos médicos e ingenieros cualificados para analizar el dispositivo explantado, revisar las imágenes médicas y establecer una conexión clara entre el fallo del producto y la lesión.
Indemnizaciones recuperables en los casos de responsabilidad por productos defectuosos en Arizona
Las víctimas de dispositivos defectuosos pueden obtener una indemnización por cirugías de revisión, salarios perdidos, daños y perjuicios por dolor y sufrimiento y, en casos de negligencia grave, daños punitivos. Las categorías de daños indemnizables suelen incluir tanto pérdidas económicas como no económicas.
Daños económicos cubrir pérdidas económicas cuantificables, como los gastos médicos derivados de cirugías de revisión y la pérdida de ingresos. Estos daños y perjuicios ayudan a los pacientes a recuperar el dinero que han gastado o perdido a causa de la lesión:
- Gastos médicos por cirugías de revisión, hospitalizaciones y tratamientos continuados
- Costos médicos futuros relacionados con el fallo del dispositivo
- Pérdida de salario y disminución de la capacidad de ganancia
Los daños no económicos se refieren al impacto personal de la lesión:
- Sufrimiento físico
- Angustia emocional
- Pérdida del disfrute de la vida y de las actividades cotidianas
Se pueden conceder indemnizaciones punitivas si las pruebas demuestran que el fabricante actuó con intención de causar daño o con un desprecio consciente por la seguridad del paciente. Estas indemnizaciones no están vinculadas a las pérdidas sufridas por el paciente, sino que tienen por objeto hacer que la empresa rinda cuentas por su conducta grave. Si el fallo de un dispositivo provoca la pérdida de un ser querido, las familias también pueden presentar una demanda por homicidio culposo para cubrir los gastos del funeral y la pérdida de compañía. Un abogado especializado en lesiones causadas por dispositivos médicos puede evaluar si los hechos de su caso respaldan estas reclamaciones.
Póngase en contacto hoy mismo con los abogados especializados en dispositivos médicos de Arizona de Hastings Law Firm para obtener ayuda
Los dispositivos médicos defectuosos pueden provocar complicaciones que alteran la vida, desde dolorosas cirugías de revisión hasta enfermedades crónicas que transforman tu vida cotidiana. Si un dispositivo falló dentro de tu cuerpo, no deberías tener que soportar esa carga tú solo, ni deberías tener que pagar por el error de otra persona.
Hastings Law Firm representa a pacientes y familias que han sufrido daños a causa de productos médicos defectuosos. Nuestro equipo cuenta con profesionales médicos internos y exabogados defensores que conocen de primera mano cómo responden los fabricantes de dispositivos y sus aseguradoras ante las reclamaciones. Tommy Hastings, un abogado litigante certificado por la junta con más de 20 años de experiencia, fundó nuestro bufete para dar voz a los pacientes que han sufrido daños a causa de errores médicos.
Nuestra Si no ganamos, no cobramos Nuestra política implica que no tendrá que pagar honorarios ni gastos de abogados a menos que consigamos una indemnización para usted. Póngase en contacto hoy mismo con nuestra oficina de Phoenix para obtener una evaluación gratuita y confidencial de su caso. Estamos aquí para ayudarle a comprender lo que ha sucedido y cuáles son los siguientes pasos.
Preguntas frecuentes sobre dispositivos médicos defectuosos en Arizona

Términos clave sobre productos sanitarios defectuosos:
- Notificación de productos sanitarios (MDR)
- Un sistema obligatorio que exige a los fabricantes, importadores y centros de salud que notifiquen a la Administración de Alimentos y Medicamentos (FDA) las lesiones graves, los fallos de funcionamiento o las muertes relacionadas con productos sanitarios. En los casos de productos defectuosos, los registros del MDR pueden revelar si el fabricante tenía conocimiento de los problemas del producto antes de que se produjera la lesión, lo cual constituye una prueba fundamental para demostrar la responsabilidad.
- Retirada de un producto sanitario
- Medida adoptada por un fabricante o por la FDA para retirar del mercado o corregir un dispositivo médico que supone un riesgo para la salud o infringe la legislación federal. Las retiradas del mercado se clasifican según su gravedad: Clase I (riesgo grave de lesiones o muerte), Clase II (problemas de salud temporales o reversibles) y Clase III (poco probable que cause daños). Una retirada del mercado no prueba automáticamente que un defecto haya causado su lesión, pero constituye una prueba sólida de que el fabricante reconoció un problema de seguridad.
- Defecto de diseño
- Un defecto inherente al plano o al diseño de un dispositivo médico que hace que toda la línea de productos sea irrazonablemente peligrosa, incluso cuando se fabrica y utiliza exactamente según lo previsto. Por ejemplo, si un implante de cadera está diseñado con componentes de metal contra metal que liberan partículas tóxicas en el cuerpo, cada unidad de ese modelo conlleva el mismo riesgo. En los casos de responsabilidad por productos defectuosos en Arizona, se puede exigir al fabricante la responsabilidad objetiva por defectos de diseño sin necesidad de demostrar que la empresa actuó con negligencia.
- Omisión de advertencias (defecto de comercialización)
- Un tipo de defecto del producto que se produce cuando un fabricante conoce o debería conocer los riesgos graves de un dispositivo médico, pero no proporciona las advertencias o instrucciones adecuadas a los médicos y pacientes. Aunque un dispositivo esté correctamente diseñado y fabricado, el fabricante puede ser considerado responsable si ocultó peligros, minimizó los efectos secundarios o no actualizó las advertencias tras tener conocimiento de nuevos riesgos. A veces se denomina «defecto de comercialización», ya que tiene que ver con la forma en que se promocionó y etiquetó el producto.
- Cirugía de revisión
- Una intervención quirúrgica de seguimiento necesaria para reparar, sustituir o extraer un dispositivo médico defectuoso que se implantó en una intervención anterior. Las cirugías de revisión suelen ser más complejas, dolorosas y arriesgadas que la cirugía de implante original. En los casos de dispositivos defectuosos, la necesidad de una cirugía de revisión —junto con los gastos médicos, el tiempo de recuperación y las complicaciones asociadas— es un componente clave de la indemnización que puede reclamar al fabricante.
- Filtro de vena cava inferior (VCI)
- Un pequeño dispositivo metálico que se implanta en la vena más grande del cuerpo (la vena cava inferior) para atrapar coágulos sanguíneos y evitar que lleguen a los pulmones. Los filtros de la vena cava inferior están diseñados para ser temporales, pero con frecuencia se dejan colocados durante demasiado tiempo, lo que provoca complicaciones graves, como la rotura del dispositivo, su desplazamiento por el cuerpo o la perforación de órganos. Estos filtros se encuentran entre los dispositivos médicos defectuosos que más litigios generan debido a sus elevadas tasas de fallo y complicaciones.
- Autorización FDA 510(k)
- Una vía de aprobación simplificada que permite que un dispositivo médico llegue al mercado demostrando que es sustancialmente similar a un dispositivo que ya se encuentra en el mercado, en lugar de demostrar su seguridad y eficacia mediante rigurosas pruebas clínicas. Muchos dispositivos de alto riesgo ingresan al mercado a través de esta laguna legal, y la autorización 510(k) no protege a los fabricantes de la responsabilidad si el dispositivo causa lesiones a los pacientes. En los litigios por dispositivos defectuosos, el hecho de que un dispositivo haya recibido solo la autorización 510(k) —y no la aprobación completa— puede constituir una prueba contundente de que nunca se probó adecuadamente.
- Aprobación previa a la comercialización (PMA)
- El tipo de proceso de revisión más riguroso de la FDA, reservado para dispositivos médicos de alto riesgo, como marcapasos y válvulas cardíacas. La aprobación previa a la comercialización (PMA) exige que los fabricantes presenten datos clínicos exhaustivos que demuestren que el dispositivo es seguro y eficaz antes de que pueda venderse. Los dispositivos que obtienen la PMA están sujetos a normas federales de prevalencia, las cuales pueden limitar —pero no eliminar— su capacidad para demandar al fabricante en virtud de la legislación estatal si el dispositivo falla y causa lesiones.
- Archivos de datos MDR | Administración de Alimentos y Medicamentos de EE.UU.
- Notificación de dispositivos médicos (MDR): cómo informar sobre problemas con dispositivos médicos | Administración de Alimentos y Medicamentos de EE.UU.
- Cómo analizar y comercializar tu dispositivo | Administración de Alimentos y Medicamentos de los Estados Unidos
- 12-542 Lesiones personales; lesiones con resultado de muerte; daños a la propiedad; apropiación indebida de bienes; allanamiento de morada y retención ilegal; plazo de prescripción de dos años | Legislatura de Arizona
Obtenga respuestas hoy mismo
Si cree que una negligencia médica, un medicamento peligroso o un producto médico defectuoso le han causado daños a usted o a un ser querido, nuestro equipo está a su disposición para ofrecerle orientación. Le explicaremos las opciones que le ofrece la legislación vigente y le ayudaremos a avanzar con claridad y comprensión. Las revisiones de casos son gratuitas y 100% confidenciales.
